
MEGA :: Molecular Evolutionary Genetics Analysis
將物種基因或蛋白變異利用此軟體分析物種之間親緣關係,並畫出phylogenetic tree
1.先下載軟體 http://www.megasoftware.net/mega.php
2.找物種,我以松科Pinaceae為範例

3.在NCBI Nucleotide搜尋 Pinaceae 及 rbcl 基因,任意選擇十個結果,儲存成 FASTA檔

4.先將軟體MEGA 打開,再開啟 FASTA檔,各物種基因序列都會匯入分析軟體
分析前有些步驟要注意,如以下
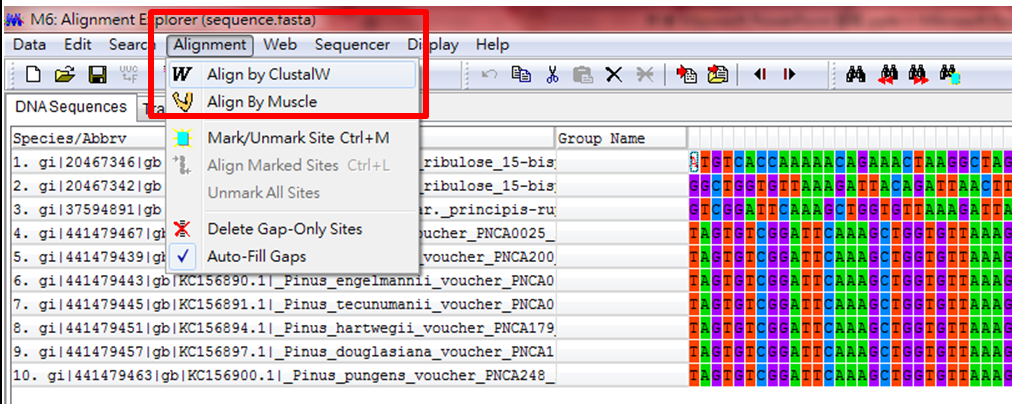
(1)先將基因序列Alignment,排列正確,兩者都可以選,資料龐大時,可以用下面那個
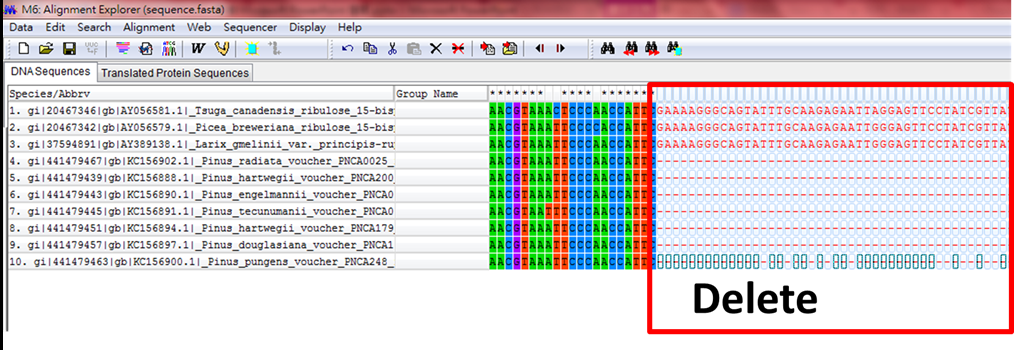
(2)將前後多出來的基因序列切齊,以方便軟體快速分析
(3)將FASTA檔轉成MEGA可以辨識的格式

出現以下,TA視窗,此時才能分析DATA
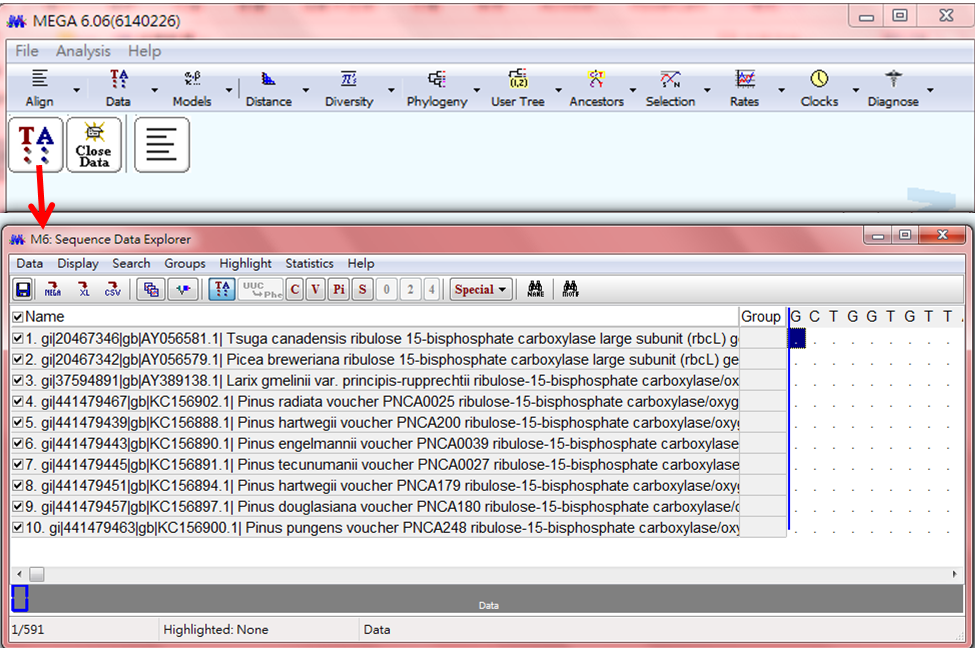
#在此可以用顏色區別功能,看出大致基因序列的差異,如以下:

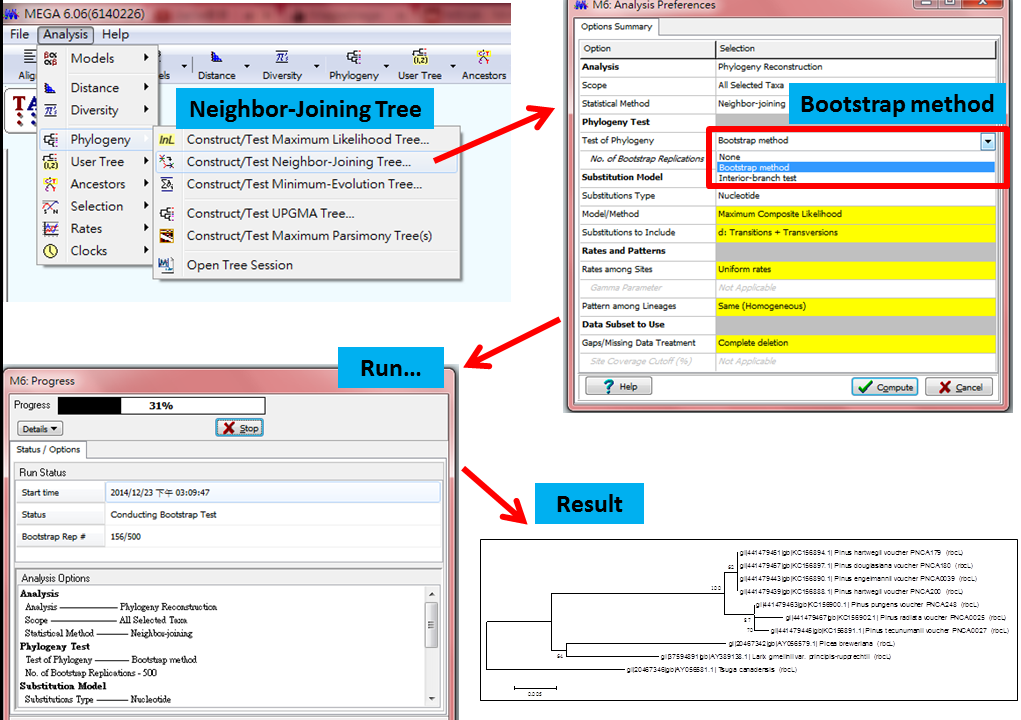
5.分析數據,一定要選Bootstrap method,為可信度的依據,步驟如圖:
Neighbor-Joining Tree 分析
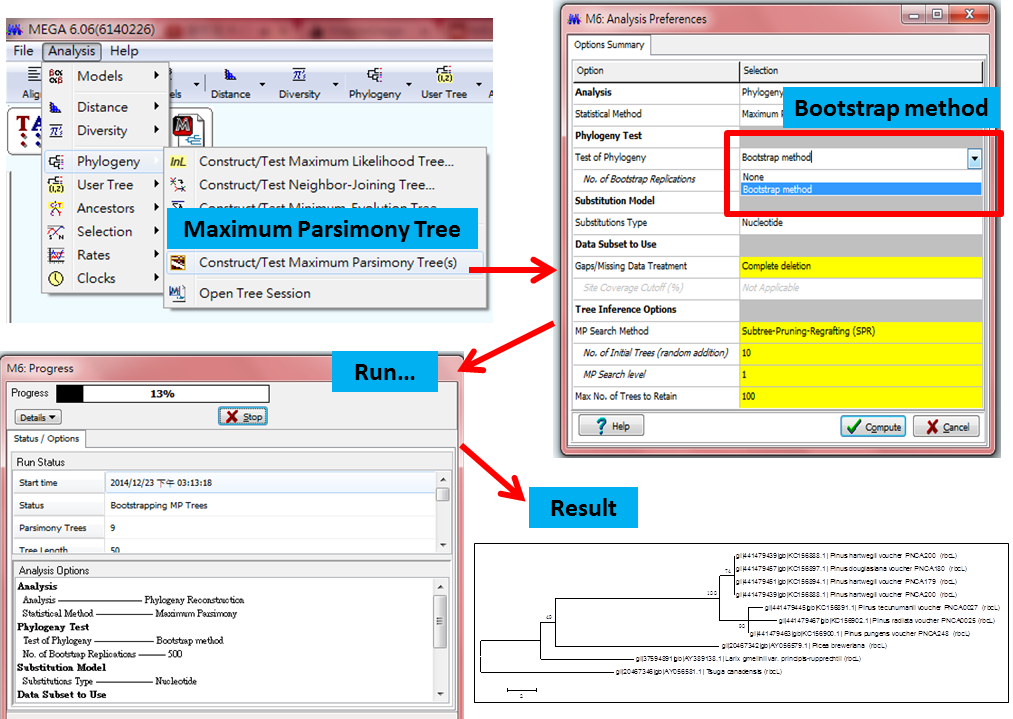
Maximum Parsimony Tree 分析
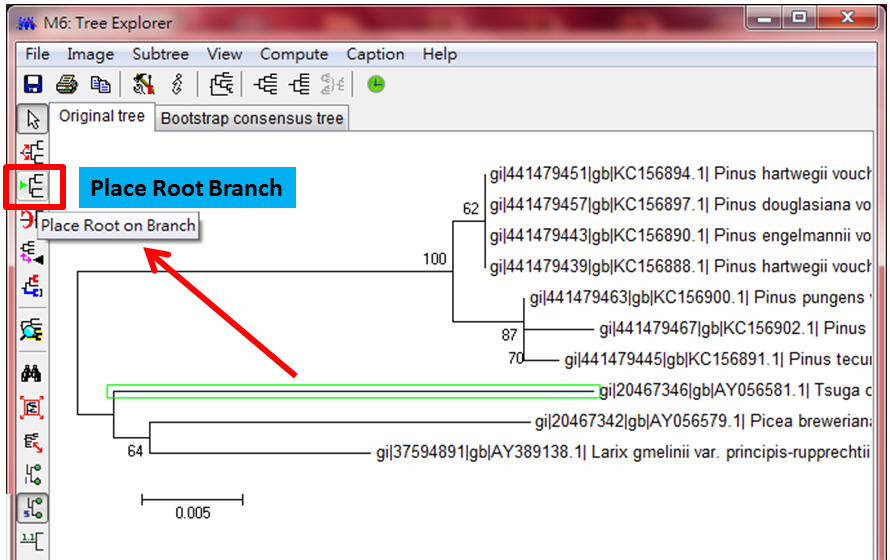
#結果要選一個物種Place Root Branch,然後可以對比這兩個結果的差異與真實性
6.結果討論
Neighbor-Joining Tree
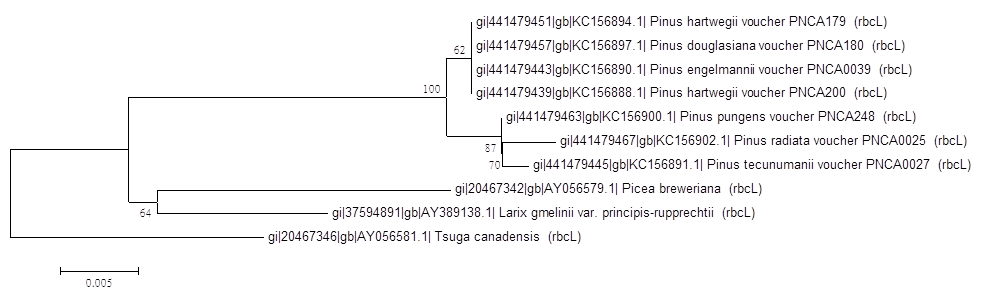
Maximum Parsimony Tree
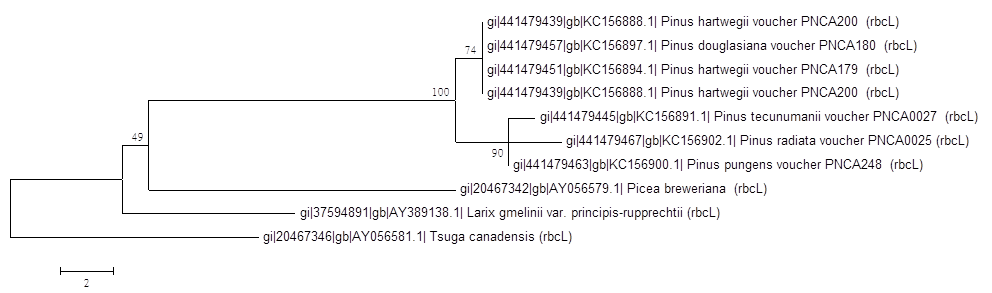
兩個結果不太一樣,就顯著差異而言,Neighbor-Joining Tree的結果 可信度較高,而Maximum Parsimony Tree顯著差異太大,不可信。但是Neighbor-Joining Tree的Bootstrap 數值並非都很高,所以物種的親緣性數據支持度並沒有很充足,還需要參考其他數據結果,不能只單看一個基因。